
Das Klinefelter-Syndrom bezeichnet die Auswirkungen einer besonderen Chromosomenkonstellation bei Jungen bzw. Männern, die zusätzlich zum normalen Chromosomensatz 46,XY, ein weiteres X-Chromosom in sich tragen. Weil sich eine breite Variabilität in der Ausprägung der Symptome findet, wird das Syndrom oft nicht erkannt.
Vom Vollbild der Klinefelter-Symptomatik betroffene Männer sind überdurchschnittlich groß und wirken besonders langbeinig und langarmig. Sie haben schmale Schultern und breite Hüften und weisen eine spärliche Körperbehaarung mit fehlendem oder vermindertem Bartwuchs auf. Sie entwickeln eine Gynäkomastie und haben eine Hodenatrophie und es kann eine Azoospermie vorliegen. Im höheren Lebensalter können diese Männer eine Skoliose sowie eine Osteoporose entwickeln. Häufig wird auch ein Diabetes mellitus beobachtet. Das Risiko für Brustkrebs ist erhöht (nur einer von 100.000 Männern erkrankt an Brustkrebs, Patienten mit Klinefelter-Syndrom haben eine 15- bis 50- fache Risikoerhöhung auf etwa 1:2000 bis 1:10000). Die Ausprägung in der Schwere der Symptomatik ist jedoch individuell sehr verschieden. Nur wenige zeigen das klinische Vollbild; meist liegt es in unterschiedlich abgeschwächter Form vor.
Häufigkeit
Einer von etwa 500-600 Männern ist Träger des 47,XXY-Karyotyps. Man nimmt an, dass nur 25 bis 30% der Patienten mit Klinefelter-Syndrom diagnostiziert und nur weniger als 10% vor der Pubertät entdeckt werden.
Karyotyp
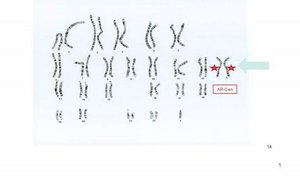
Das Syndrom wird nachgewiesen durch ein überzähliges X-Chromosom in der Chromosomenanalyse: Karyotyp 47,XXY. Normalerweise liegen 46 Chromosomen vor. 22 Autosomen und 2 Geschlechtschromosomen, X- und Y-Chromosom, wobei durch das SRY-Gen auf dem Y-Chromosom das männliche Geschlecht determiniert wird. 90% aller Träger des Klinefelter-Syndroms zeigen den Karyotyp 47,XXY; 7% der Patienten weisen Mosaikformen auf wie z. B. 47,XXY/46,XY. Klar abzutrennen vom klassischen Klinefelter-Syndrom sind Karyotyp-Varianten mit weiteren überzähligen X- und Y-Chromosomen (z.B. 48,XXXY oder 49,XXXXY, 49,XXXYY). Bei diesen Patienten liegen zusätzlich eine geistige Retardierung und weitere Fehlbildungen vor.
Erstbeschreibung
Der amerikanische Arzt H.F. Klinefelter veröffentlichte 1942 in Boston einen Aufsatz, in dem er das Syndrom erstmals beschrieben hatte. 1950 entdeckte M.L. Barr bei Klinefelter-Patienten ein kondensiertes Kernkörperchen in den Zellen, das sogenannte Barrkörperchen. Dieses Barrkörperchen stellte sich später als inaktiviertes X-Chromosom heraus. Erst in den späten Fünfziger Jahren wurde von P.A. Jacobs und J.A. Strong die genetische Ursache in Form eines zusätzlichen X-Chromosoms durch Analyse von Chromosomenpräparaten aus Knochenmarkszellen entdeckt.
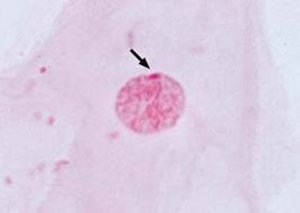
Abb. 2: Barr-Körperchen
Entstehung
Diese numerische Chromosomenaberration entsteht durch Fehlverteilung der Chromosomen während der Keimzellbildung, also der Ei- oder Spermienbildung. In 2/3 der Fälle kommt das überzählige X-Chromosom von der Mutter. Die Häufigkeit der mütterlichen Fehlverteilungen in den meiotischen Teilungen nimmt mit dem Alter der Mutter zu. Für die restlichen Fälle konnte eine väterliche Herkunft nachgewiesen werden.
Entwicklung
Kindesalter
Manchmal sind die Jungen mit Klinefelter-Syndrom etwas entwicklungsverzögert, zeigen eine Sprachschwäche und haben Konzentrations- und Kontaktschwierigkeiten. Diagnostisch hinweisend können auch extremitätenbetonter Hochwuchs oder Hodenhochstand sein. Intellektuell scheinen sie 5 bis 10 Prozentpunkte unter dem Intelligenzquotienten (eher bezogen auf den sprachlichen IQ) der Geschwister zu liegen. Unterstützend sollten also bei Bedarf Sprach- und Bewegungstherapien angeboten werden.
Pubertät
Meist fallen die vom Klinefelter-Syndrom betroffenen Jungen erst in der Pubertät auf, wenn die Ausbildung der sekundären Geschlechtsmerkmale ausbleibt, da in den unterentwickelten Hoden zu wenig Testosteron gebildet wird. Achsel-, Scham-, und Körperbehaarung sind ebenso wie der Bartwuchs stark vermindert. Bei Hormonanalysen finden sich niedrige Androgenwerte, sowie hohe Gonadotropinwerte (LH und FSH) und hohe Estradiolwerte. Ein Klinefelter-Patient sollte etwa im Alter von 11 Jahren einem Kinderendokrinologen vorgestellt werden, damit der richtige Zeitpunkt für den Start der Therapie gewählt werden kann.
Therapie
Ab der Pubertät sollte eine Testosteron-Therapie in Form von Pflastern, Depotspritzen oder Gels erwogen werden. Nicht alle Patienten brauchen eine Hormontherapie. In Abhängigkeit der Ausprägung des Testosteronmangels wird die Therapie oft erst im Erwachsenenalter nötig. Es wird individuell, je nach klinischem Bild und Hormonwerten, die jeweilige Dosis (auch unterschiedlich im zeitlichen Verlauf) bestimmt. Diese Substitution erfolgt dann meist ein Leben lang.Die Testosterongabe bewirkt die Vermännlichung des Körperbaus, die Bildung von Körperbehaarung und des Bartwuchses, das Tieferwerden der Stimme und das Wachstum des Penis. Das sexuelle Verlangen wird dadurch angeregt. Die Wachstumsfugen schließen sich, wodurch das Längenwachstum beendet wird. Unter der Testosterontherapie sind fortlaufende Kontrollen des Blutbildes, der Leberenzyme, sowie die Kontrolle auf Prostatakarzinom und auf Brustkrebs erforderlich. Vor der Hormon-Substitution kann eine genetische Untersuchung des CAG-Repeat Polymorphismus im AR-Gen durchgeführt werden. Hier wird der Androgen-Rezeptor-Typ bestimmt. Bei kurzen Repeats (unter 20) ist die Testosteron/Androgenwirkung besser, da auf eine Androgentherapie besser angesprochen wird. Bei längeren Repeats ist die Androgen-Aktivität erniedrigt, sodass die medikamentöse Therapie mit Testosteron nicht gut wirkt (eventuell muss dann die Dosis erhöht werden).
Kinderwunsch
Früher galten Männer mit Klinefelter-Syndrom als zeugungsunfähig, da sie wenig bis gar keine Spermien produzieren. Nur bei etwa der Hälfte der Patienten finden sich Spermien im Hodenbiopsat. Eine Hormontherapie hat leider zur Folge, dass bei diesen Männern die Gewinnung von Spermien weniger gut gelingt als bei unbehandelten Patienten. Es ist daher empfehlenswert, vor einer Testosterontherapie Spermien zu asservieren. Zu Beginn der Pubertät gibt es ein Zeitfenster mit zunächst steigenden Testosteronwerten. Hier scheint der beste Zeitpunkt zu sein, Spermien im Ejakulat oder durch Hodenbiopsie zu gewinnen, um sie für eine spätere Sterilitätsbehandlung ein- zufrieren (Kryokonservierung). Mittlerweile können Klinefelter-Patienten mit den Techniken der modernen Reproduktionsmedizin möglicherweise Vater werden. Hierbei kommen zwei Methoden, die Intracytoplasmatische Spermieninjektion (ICSI) und die Testikuläre Spermienextraktion (TESE) zur Anwendung. Nach einer Hormonbehandlung der Frau werden die reifen Eizellen entnommen. Die bei der Hodenbiopsie gewonnenen Spermien werden in vitro in die Eizellen injiziert. Die Erfolgsquote nimmt für Männer über 35 Jahre rapide ab, weswegen eine Fertilisationsbehandlung besser in jüngeren Jahren erfolgen sollte. Die erzielten Schwangerschaftsraten unterscheiden sich nicht von denen der Kontrollgruppe. Mehrheitlich hatten die Kinder nach dieser Behandlung normale Karyotypen. Es gab aber auch Söhne, die wiederum ein Klinefelter- Syndrom zeigten oder Mädchen, die ein Triple-X- Syndrom aufwiesen.
Genetische Beratung
In einem persönlichen Gespräch werden genetische Ursachen, Vererbungsmuster, Möglichkeiten der Diagnostik, Therapie und Prognose erörtert. Werdende Eltern, die einen Sohn mit Klinefelter- Syndrom erwarten, oder Eltern, bei deren Kindern der Verdacht auf ein Klinefelter-Syndrom geäußert wurde, bzw. Betroffene im Erwachsenenalter können sich an die Beratungsstellen des genetikum wenden.
Selbsthilfegruppe
Es steht auch eine deutsche Selbsthilfegruppe zur Verfügung, zu finden unter www.klinefelter.de. Bernhard Köpl, erster Vorstand, setzt sich für einen offenen Umgang mit dem Syndrom ein: „Das Klinefelter-Syndrom ist viel zu wenig bekannt. Ich rate allen Betroffenen ganz offen damit umzugehen und sich an unsere Selbsthilfegruppe zu wenden, denn man kann sich in die Problematik überhaupt nicht rein denken, wenn man das Klinefelter-Syndrom nicht hat.“ Es finden regelmäßige Treffen der Selbsthilfegruppe statt.
Telefonkontakt: 0049 176 503 902 56, Fax: 0049 3212 1573345.
47xxy klinefelter syndrom e. v. // Geschäftsstelle: Hauzenberger Str. 32, 80687 München
Vorstand: Bernhard Köpl, Dirk Mueller, Christian Uhlenbroich
VR Düsseldorf Nr. 10944 St.-Nr. 107/5703/3042