Erbliche Krebserkrankungen kommen sehr viel häufiger vor als bis vor kurzem noch angenommen. Welche Tumorsyndrome können genetisch bedingt sein? Und warum ist eine genetische Beratung dann wichtig?
In Deutschland treten etwa 500 000 Neuerkrankungen an Krebs pro Jahr auf. In etwa fünf Prozent der Fälle muss aufgrund einer bestehenden familiären Häufung an eine erbliche Disposition gedacht werden. Bei dieser großen Zahl von Patienten – sie ist natürlich noch viel größer, wenn man die nicht erkrankten, aber gefährdeten Familienmitglieder dazuzählt – ist es unbedingt notwendig zu prüfen, bei welchen Personen eine genetische Beratung und/ oder Diagnostik sinnvoll wäre.
Jede Form von Krebs beruht auf genetischen bzw. epigenetischen Veränderungen der Tumorzellen. Dabei ist es nicht nur eine Mutation, sondern eine Vielzahl von Mutationen in verschiedenen Genen, die aus einer normalen Zelle eine maligne und invasiv wachsende Tumorzelle machen. Die modernen DNA-Sequenzierungstechniken haben es erlaubt, ganze Genome von Krebszellen zu analysieren. Die Zahl der so gefundenen Mutationen geht in die Hunderte. Dabei handelt es sich um somatische Mutationen, die nur in den Tumorzellen selber vorliegen. Bei erblichen Krebserkrankungen liegt von Beginn an in allen Zellen des Individuums eine Mutation in einem Krebsgen (häufig in einem Tumorsuppressorgen) als Keimbahnmutation vor. Nach einem zufälligen Prozess wird mit hoher Wahrscheinlichkeit auch die zweite, bisher intakte Kopie eines solchen Tumorsuppressorgens im Laufe des Lebens durch Mutationen verändert. Dann hat diese Zelle ein hohes Potential zu entarten. Aus bisher wenig verstandenen Gründen sind diese Krebsgene jeweils für die Entwicklung eines definierten Spektrums von Tumoren verantwortlich, z. B. bei den Genen BRCA1 und BRCA2 insbesondere für Brust- und Eierstockkrebs. Als Anzeichen für ein genetisches Tumorsyndrom gelten daher: ein definiertes Spek- trum von Tumoren bei direkten Verwandten und ein deutlich jüngeres Ersterkrankungsalter (mind. ein Patient unter 50 Jahren). Informativ für eine genetische Untersuchung sind die Merkmalsträger selber, d. h. die Patienten, die einen Tumor entwickelt haben. Die genetische Untersuchung ist aus zwei Gründen sinnvoll: Sie erlaubt eine Risikovorhersage für die direkten Verwandten, und sie ist hilfreich für die Wahl der geeigneten Therapie für den Patienten selbst.
Tumore, bei denen Gene Auslöser sein können
Das Retinoblastom ist das Lehrbuchbeispiel für ein genetisches Tumorsyndrom. Bei etwa einem Drittel der betroffenen Patienten liegt eine Keimbahnmutation im entsprechenden RB1-Gen vor. Häufig erkranken Kinder sehr früh an diesem Tumor, teilweise ist er sogar embryonal angelegt. Bei etwa 90 Prozent der Personen mit einer Keimbahnmutation im RB1-Gen tritt in einer der Retinazellen spontan eine zweite Mutation auf, und es entwickelt sich ein Tumor. Neben Tumoren der Retina besteht für diese Patienten später ein Risiko für Osteo- bzw. Chondrosarkome und Melanome.
Brust- und Eierstockkrebs: In etwa 10 Prozent dieser verbreiteten Erkrankung – etwa jede 12. Frau erkrankt daran – wird eine familiäre Häufung festgestellt. In einem kleineren Teil der betroffenen Familien kann eine Mutation in einem von den beiden Brustkrebsgenen (BRCA1 und BRCA2) nachgewiesen werden. Damit ist die Frequenz dieser Mutationen in unserer Bevölkerung relativ hoch (etwa 1:400 bis 1:500, sowohl für Frauen als auch für Männer). Es handelt sich bei BRCA1 und BRCA2 um DNA- Reparaturgene, die aus nicht verstandenen Gründen zur Krebsentstehung insbesondere in der weiblichen Brustdrüse und in den Eierstöcken führen. Tumore in anderen Organen treten mit einer deutlich ge- ringeren Frequenz auf (Tab. 1), hier können dann auch Männer betroffen sein. Eine spezielle Tumor- vorsorge betrifft bisher nur den Brust- und Eierstockkrebs. Seit mehr als 15 Jahren wird versucht, in entsprechenden Familien weitere, relativ häufige Brustkrebsgene zu identifizieren, jedoch bisher ohne Erfolg. Für Brustkrebspatienten hat der Mutationsnachweis Bedeutung für die einzuschlagende Therapie. Bislang gesunden und bereits erkrankten Frauen mit einer BRCA1- oder BRCA2-Mutation werden intensivierte Früherkennungs-Programme angeboten. Zudem kann das Tumorrisiko durch die prophylaktische operative Entfernung von Brust und Eierstöcken nach der Reproduktionsphase deutlich gesenkt werden.

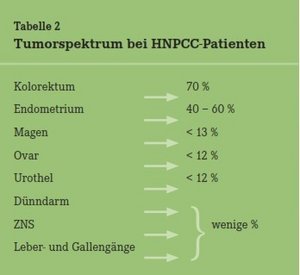
Dickdarmkrebs: Diese Krebsform steht bei Männern und Frauen nach Prostata- bzw. Brustkrebs an zweiter Stelle der Krebserkrankungen. Von den etwa 75 000 Neuerkrankungen an Dickdarmkrebs pro Jahr in der Bundesrepublik liegt bei etwa drei Prozent eine vererbte Tumordisposition vor. Daraus errechnet sich eine Häufigkeit von etwa 1:500 von entsprechenden Genträgern bzw. Risikopatienten in unserer Bevölkerung. Damit ist das Dickdarmkarzinom das häufigste genetische Tumorsyndrom. Bei einzeln wachsenden Tumoren (also nicht Poly- posis) handelt es sich fast immer um das HNPCC- Syndrom (hereditary non-polyposis colon cancer). Verantwortlich sind Mutationen in einem von vier DNA-Reparaturgenen, und auch hier wird wieder ein bestimmtes Spektrum von Tumoren (Tab. 2) induziert. Das Risiko für eine Tumorentstehung ist bei dieser Disposition hoch, bleibt aber immer unter 100 Prozent. Nicht erkrankten Mutationsträgern werden frühzeitig regelmäßige Vorsorgeuntersuchungen empfohlen. Diese sind sehr effektiv, weil diese Tumoren erst relativ spät metastasieren.
Neben dem HNPCC-Syndrom gibt es eine Vielzahl von sehr viel selteneren Syndromen, bei denen auch Dickdarmtumore im Vordergrund stehen. Darunter ist auch die familiäre adenomatöse Polyposis (FAP): Hier ist oft schon bei Jugendlichen der Dickdarm mit Polypen übersät. Aus diesen Polypen entwickelt sich mit einer Wahrscheinlichkeit von 100 Prozent sehr früh ein Karzinom. In diesen seltenen Fällen sollten in den entsprechenden Familien bereits Kinder auf die Anlageträgerschaft untersucht werden, unter Umständen muss ab dem 10. Lebensjahr mit Vorsorgeuntersuchungen begonnen werden.
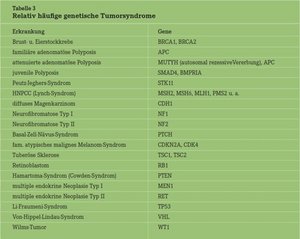
Es gibt eine ganze Liste von weiteren Tumorsyndromen (Tab. 3), für die jeweils ein spezifisches Spektrum von Tumoren charakteristisch ist. Allen gemeinsam sind die autosomal-dominante Vererbung und das jeweilige hohe spezifische Tumorrisiko. Jeder genetische Berater hat schon vereinzelt Familien mit diesen hohen Tumorrisiken erlebt. Er sieht sich dann Menschen gegenüber, die sich unmittelbar schicksalhaft bedroht fühlen. Ihren Ängsten können wir Mediziner mit unseren einfachen, sehr naturwissenschaftlichen Begründungen nie gerecht werden. Die psychoonkologische Betreuung ist deshalb fester Bestandteil der Beratung.