
Die erblich bedingten Myotonien sind eine Gruppe von Muskelerkrankungen, die durch verzögerte Muskelerschlaffung charakterisiert sind. Klinisch werden sie in der Regel in zwei Gruppen eingeteilt: die myotonen Dystrophien und die nicht-dystrophen Myotonien. Die nicht-dystrophen Myotonien werden durch Mutationen in denjenigen Genen verursacht, die die Information für die muskulären Kanalproteine tragen („Kanalopathien“).
Bei den myotonen Dystrophien handelt es sich um Multisystemerkrankungen, bei denen neben den muskulären Symptomen auch andere Organe betroffen sind. Dabei wird zwischen zwei Typen unterschieden, die beide autosomal dominant vererbt werden: der Myotonen Dystrophie Typ 1 (DM1, Dystrophia myotonica Steinert) und der Myotonen Dystrophie Typ 2 (DM2, proximale myotone Myopathie, PROMM). Die Häufigkeit beider Erkrankungen wird in Deutschland auf etwa 1:10.000 – 1:15.000 geschätzt. Damit zählt die Gruppe myotoner Dystrophien zu den beim Erwachsenen am häufigsten vorkommenden Muskelerkrankungen unseres Kulturkreises. Es wird vermutet, dass DM1 und DM2 in den Ländern außerhalb Mitteleuropas deutlich seltener vorkommen.
Motone Dystrophie Typ 1 (Curschmann-Steinert-Syndrom)
Die myotone Dystrophie Typ 1 (OMIM #160900) wurde 1909 klinisch von Steinert beschrieben (vgl. Steinert). Neben der bereits genannten Muskelsteifheit (Myotonie) treten als weitere muskuläre Symptome vor allem Muskelschwäche und Muskelatrophie in den Händen und Beinen sowie eine Veränderung der Gesichtsmuskulatur auf, die auch als „Facies myotonica“ bezeichnet wird. Zu den nichtmuskulären Symptomen der DM1 zählen in erster Linie die Linsentrübung (Katarakt oder „grauer Star“), Herzproblematiken (Kardiomyopathie und Reizleitungsstörungen) sowie endokrine Störungen wie beispielsweise ein Hypogonadismus (Mangel an Geschlechtshormonen), der vorwiegend bei Männern auftritt. In einigen Fällen kann auch ein Diabetes mellitus auftreten.
Des Weiteren können Hautveränderungen, reduzierte Funktionen innerhalb des Immunsystems und Beeinträchtigungen der geistigen Fähigkeiten hinzukommen. Aufgrund der enormen phänotypischen Variabilität der DM1 werden in der Regel drei klinische Formen zur Beschreibung des Krankheitsbildes unterschieden:
- die adulte Form, die sich in der Regel erst im hohen Alter entwickelt und sich vorwiegend durch eine Linsentrübung und eine Ptosis äußert,
- die klassische Form, die im frühen Erwachsenenalter auftritt und durch Myotonie, „Facies myotonica“ und die beschriebene extramuskuläre Symptomatik charakterisiert ist,
- die kongenitale Form, die sich teilweise bereits intrauterin manifestiert. Diese Form geht nach der Geburt meist mit schweren Muskelhypotonien („floppy infant“ und zeltförmig offenstehender Mund) sowie geistigen Beeinträchtigungen einher. Dabei tritt die kongenitale Form der DM1 fast ausschließlich dann auf, wenn das verlängerte Repeat von der Mutter stammt.
Erst 1992, ein Jahr nach der Entdeckung des neuen Mutationstyps „Trinukleotid-Repeat-Expansion“ beim fragilen X-Syndrom, konnte die genetische Ursache der DM1 identifiziert werden. Sie liegt in einer abnormen Verlängerung des CTG-Repeats in der 3‘ untranslierten Region im letzten Exondes DMPK-Gens (Dystrophia myotonica Proteinkinase), welches sich auf dem langen Arm des 19. Chromosoms befindet (vgl. Brook et al.). Während sich bei gesunden Kontrollpersonen 5–37 CTG-Einheiten im DMPK-Gen nachweisen lassen, ist die Anzahl der CTG-Repeats bei Betroffenen viel höher: Sie weisen mindestens 50 CTG-Repeats auf, wobei in einigen Fällen sogar Repeatlängen von bis zu etwa 4000 beschrieben wurden. Aufgrund der dominanten Vererbungsweise liegt das Erkrankungsrisiko für die Nachkommen von Betroffenen bei 50 %. Bei der Vererbung ist das Phänomen der klinischen Antizipation seit langem bekannt: So tritt die Erkrankung von Generation zu Generation früher auf und erschwert sich zunehmend. Dabei besteht eine gewisse Korrelation zwischen der Repeatlänge und der klinischen Ausprägung der Erkrankung. Bei Patienten, die im Erwachsenenalter erkranken, findet sich meist ein kurzes CTG-Repeat, wohingegen sich bei kongenital betroffenen Kindern in der Regel ein Repeat mit über 500 CTG-Einheiten nachweisen lässt. Da die Übergänge fließend sind, können keine sicheren prädiktiven Aussagen bezüglich der klinischen Form getroffen werden, insbesondere nicht im Rahmen einer Pränataldiagnostik.
Myotone Dystrophie Typ 2 (PROMM)
Die Myotone Dystrophie Typ 2 (OMIM #116955) wurde 1994 von Ricker und Kollegen beschrieben (vgl. Ricker et al.). Da die Muskelschwäche vorwiegend in der proximalen Muskulatur wie Oberarm und Oberschenkel auftrifft, wird die Erkrankung auch als PROMM (Proximale Myotone Myopathie) bezeichnet. Die klinischen Symptome der myotonen Dystrophie Typ 2 überschneiden sich in vielerlei Hinsicht mit denen der myotonen Dystrophie Typ 1; allerdings ist die DM2 im Vergleich zur DM1 normalerweise geringer ausgeprägt und betrifft vor allem Hände und Beine. Die Muskelschwäche kann sich im Verlauf der Erkrankung, wie bei DM1, auch auf die Gesichtsmuskulatur sowie die distalen Muskeln ausweiten. Eine Muskelatrophie ist typischerweise im Bereich der Oberschenkel - sowie der Schulter- und Oberarmmuskulatur zu finden. Muskelschmerzen treten bei Patienten mit einer DM2 wesentlich häufiger spontan oder nach Belastung auf. Sie können unter Umständen sehr intensiv sein und unterschiedlich lange andauern. Die nicht-muskulären Symptome wie Katarakt, Kardiomyopathie, Hypogonadismus und Diabetes mellitus sind im Vergleich zu DM1 ebenfalls meist geringer ausgeprägt. Vermehrtes Schwitzen und eine Wadenhypertrophie treten hingegen ausschließlich bei der DM2 auf. Die der DM2 zugrundeliegende genetische Veränderung konnte im Jahre 2001 identifiziert werden (vgl. Liquori et al.). Dabei handelt es sich um ein abnorm verlängertes CCTG-Repeat im Intron 1 des ZNF9-Gens (zinc finger protein 9) auf dem langen Arm von Chromosom 3. Während gesunde Personen bis zu 27 CCTG-Kopien auf einem ZNF9-Allel tragen, lassen sich bei DM2-Patienten etwa 75 bis über 10000 CCTG-Einheiten auf einem expandierten ZNF9-Allel nachweisen. Eine wesentliche Eigenschaft der DM2 ist, dass es nach heutigem Kenntnisstand keine konkreten Hinweise für eine kongenitale Form gibt. Darüber hinaus ist bei diesem Typ das Phänomen der klinischen Antizipation kaum erkennbar.
Genetische Instabilität bei Myotoner Dystrophie Typ 1 und Typ 2
Das Phänomen der klinischen Antizipation lässt sich anhand der genetischen Antizipation erklären, d. h. durch das Vorliegen eines Repeats in veränderter Länge bei den Nachkommen (siehe Abb. 1). Denn CTG-Repeats bzw. CCTG-Repeats, die nicht mehr im Normbereich liegen, werden instabil. Dies führt dazu, dass sich das expandierte Repeat im Zuge der Weitergabe an die nächste Generation noch weiter verlängert. Während diese genetische Antizipation sowohl bei der DM1 als auch bei der DM2 sehr ausgeprägt ist, lässt sich die klinische Antizipation hingegen praktisch nur bei der DM1 beobachten. Auf ähnliche Art und Weise geht auch der beiden Erkrankungen zugrundeliegende Pathomechanismus vonstatten. Hierbei werden die abnormen Repeat-Expansionen beim Ablesen des Gens mit in die RNA übernommen. Aufgrund der fehlerhaft expandierten Repeatanteile entstehen Sekundärstrukturen der RNA, an die sich im Zellkern bestimmte Proteine binden. Hierdurch wird die RNA unlöslich und anschließend in Einschlusskörpern vorwiegend im Zellkern angereichert. Dies ist die ausschlaggebende Ursache für das weiterhin fehlgesteuerte Spleißen der alternativen RNAs und somit für das Auftreten organspezifischer Fehlfunktionen.
Differentialdiagnostische Aspekte
Um einen klinischen Nachweis der myotonen Entladungsserien zu erbringen, gilt die EMG-Untersuchung als wegweisend. Eine augenärztliche Untersuchung auf Linsentrübung kann häufig ebenfalls hilfreich sein. Eine myotone Reaktion wird bei einigen Patienten mit einer klassischen Form der DM1 bereits durch „Händeschütteln“ oder provozierten „Lidschlag“ offensichtlich.
Bei Verdacht auf DM2 sollte aus differentialdiagnostischer Sicht auch eine nicht-genetisch bedingte Polymyositis in Betracht gezogen werden. Auch andere genetisch bedingte Krankheiten, wie die adulte Form des Morbus Pompe oder eine Gliedergürtel-Muskeldystrophie müssen in Betracht gezogen werden. Andere seltene Myopathien, wie beispielsweise eine Einschlusskörper-Myopathie oder die Mitochondriopathien, weisen zum Teil ebenfalls Symptome einer DM1 bzw. einer DM2 auf, werden aber in der Regel erst nach Ausschluss der häufigeren DM1 bzw. DM2 analysiert. Für die kongenitale Form der DM1, insbesondere im Hinblick auf Neugeborene mit einer „floppy infant“-Symptomatik, sollte auch das Prader-Willi-Syndrom oder eine Spinale Muskelatrophie in Betracht gezogen werden.
Molekulargenetische Diagnostik der DM1 und der DM2
Beide Erkrankungsformen lassen sich durch eine DNA-Analyse diagnostizieren. Eine Chromosomenanalyse ist hierbei nicht indiziert, da diese Erkrankungen zytogenetisch nicht nachzuweisen sind. Aufgrund des Gendiagnostikgesetzes, welches zum 01.02.2010 in Kraft getreten ist, muss der Patient vor seiner schriftlichen Einwilligung über Wesen, Bedeutung und Tragweite der durchzuführenden genetischen Untersuchung aufgeklärt werden. Bei einer prädiktiven Diagnostik, d. h. wenn der Ratsuchende keine Symptome der Erkrankung aufweist, hat vor der genetischen Untersuchung zunächst eine humangenetische Beratung zu erfolgen. Bei beiden Erkrankungen kann die Verdachtsdiagnose in einigen Fällen bereits durch eine PCR-Analyse verworfen werden, sofern jeweils zwei Repeats im Normbereich nachgewiesen werden können. Ist dies nicht möglich, ist eine erweiterte Analyse zum Ausschluss oder Nachweis eines expandierten Repeats durchzuführen. Hierzu ist entweder den Einsatz einer speziellen PCR- (Triple- oder Long-Range-PCR) oder einer Southern-Analyse erforderlich, die zudem auch eine Abschätzung der Repeatlänge großer Expansionen zulässt (siehe Abb. 1).
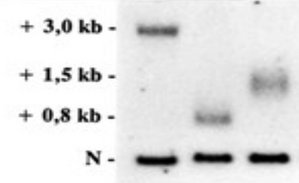
Abbildung 1: Die Southern-Analyse zeigt die genetische Antizipation. In den Folgegenerationen konnte jeweils eine deutliche Expansion des CTG-Repeats nachgewiesen werden. Die mit N bezeichneten Banden entsprechen dem Repeat des normalen DMPK-Allels. Die DNA der Großmutter (mittlere Spur) weist zusätzlich ein DMPK-Allel mit etwa 250 CTG- Einheiten auf. Bei der Mutter des betroffenen Kindes ist das Repeat bereits auf etwa 500 CTG-Einheiten angewachsen, beim Kind auf etwa 1000 CTG-Einheiten. Die Großmutter weist eine sehr milde adulte Form der DM1 auf, die sich praktisch nur auf eine Linsentrübung beschränkt. Ihre Tochter zeigt eine klassische Form der DM1 mit einer deutlichen Ptosis sowie einer Myotonie und das Kind ist von einer schweren kongenitalen Form der DM1 betroffen.