
Kasuistik: Angeborene Tibia-Pseudarthrose und Café-au-lait Flecken – gibt es einen genetischen Zusammenhang?
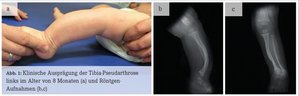
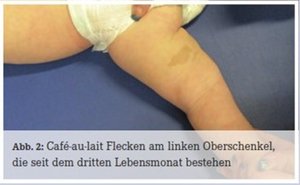
Bei Leon fiel direkt nach der Geburt ein verformter linker Unterschenkel auf. In der Röntgen-Untersuchung des betroffenen Unterschenkels stellte sich eine Tibia-Pseudarthrose links heraus (Abb. 1). Leon wurde mit einer Unterschenkel-Fuß-Orthese links versorgt. Im Alter von drei Monaten zeigten sich zwei Milchkaffee-Flecken (Café-au-lait Flecken) an der Hinterseite seines linken Oberschenkels, einer davon nahm bis zum Alter von 8 Monaten deutlich an Größe zu (Abb. 2). Im Lauf von Leons erstem Lebensjahr erschienen an Bauch und Rücken viele weitere kleine Café-au-lait Flecken. Somit waren die klinischen Kriterien für eine Neurofibromatose Typ 1 (NF1) bei Leon erfüllt und die Diagnose wurde durch den Nachweis einer Mutation im NF1-Gen (Splice-Akzeptor- Mutation c.3975-1G>C) bestätigt.
Die Familienanamnese war unauffällig. Im Blut der Eltern gab es keinen Hinweis auf dieselbe NF1-Mutation. Da die Mutation bei Leon also neu aufgetreten ist, haben die Eltern nur ein geringfügig erhöhtes Risiko, dass weitere Kinder ebenfalls betroffen sein werden. Diese geringfügige Risikoerhöhung gegenüber der Elternteil ein sogenanntes Keimzell-Mosaik vorliegen könnte. In diesem Fall findet sich die NF1-Mutation in mehreren Eizellen oder Spermien. Zugleich liegen jedoch auch Keimzellen ohne NF1-Mutation vor, daher die Bezeichnung als Keimzell-Mosaik. Im Blut der Eltern sind Keimzell-Mosaike nicht nachweisbar.
Leons Eltern wurden ausführlich beraten und traten einer Selbsthilfegruppe bei. Im zweiten und dritten Lebensjahr entwickelten sich bei Leon viele weitere Café-au-lait Flecken sowie ein inguinales Freckling. Bei einer röntgenologischen Verlaufsuntersuchung zeigte sich eine deutliche knöcherne Durchbauung am Übergang vom proximalen zum mittleren Tibiadrittel bei stabilem Antekurvations-Winkel von Fibula und Tibia. Leon lernte das freie Laufen im Alter von 18 Monaten und trägt durchgehend eine stabilisierende Unterschenkel-Fuß-Orthese, um eine Fraktur im Bereich der Tibia-Pseudarthrose zu verhindern. In orthopädischen Verlaufsuntersuchungen wird entschieden, ob und zu welchem Zeitpunkt eine Operation der Tibia-Pseudarthrose notwendig wird.
Leons Kopfumfang wuchs entlang der 97. Percentile. Seine Sprachentwicklung verlief verzögert, so dass Logopädie verordnet wurde. Bei der letzten klinischen Untersuchung im Alter von 2 7/12 Jahren zeigten sich keine Neurofibrome. Die cranielle MRT ergab keinen Hinweis auf ein Optikusgliom. Zudem waren in der augenärztlichen Spaltlampen-Untersuchung keine Lisch-Knötchen zu erkennen.
Häufigkeit
Bei der Neurofibromatose Typ 1 (NF1) handelt es sich um eine erbliche Erkrankung, die vor allem die Haut und das Nervensystem betrifft. Sie gehört daher zu den neurokutanen Erkrankungen (Phakomatosen) und wird nach ihrem Erstbeschreiber auch als Morbus von Recklinghausen bezeichnet. Die Häufigkeit wird auf 1 zu 3.000 geschätzt.
Klinische Symptomatik
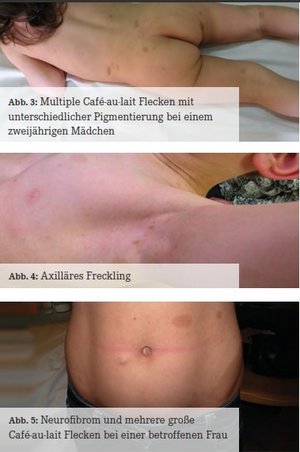
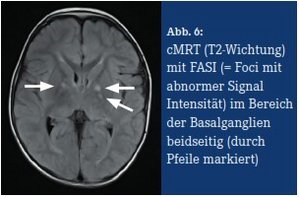
Die typischen und meist ersten Symptome sind Café-au-lait Flecken – harmlose Milchkaffeefarbige Hyperpigmentierungen im Niveau der Haut. Sie können bereits angeboren vorliegen oder sie entwickeln sich innerhalb der ersten Lebensjahre; häufig vergrößern und vermehren sie sich im Laufe der Zeit und können dunkler werden (Abb. 3). Ein weiteres, ebenfalls harmloses Hautsymptom ist eine Sommersprossen-artige Hyperpigmentierung der Achselhöhlen und Leistenregion (axilläres und inguinales Freckling, Abb. 4). Ein drittes harmloses Symptom sind Lisch-Knötchen – gutartige, weißlich aussehende Hamartome der Iris, die teilweise nur bei einer Spaltlampen-Untersuchung gesehen werden können. Meist erst im Jugend- oder Erwachsenenalter treten kutane und subkutane Neurofibrome auf (Abb. 5). Diese gutartigen Tumoren gehen von Zellen der Nervenhüllen (Schwann‘sche Zellen) aus. Eine operative Entfernung kann bei ungünstiger Lokalisation (z. B. in Gelenk-Nähe mit schmerzhafter Bewegungseinschränkung) oder aus kosmetischen Gründen angezeigt sein. Die meisten Neurofibrome bedürfen jedoch keiner operativen Entfernung. Manche NF1- Betroffene entwickeln netzartig wachsende (plexiforme) Neurofibrome, die in etwa 10 % bösartig entarten. Weitere seltenere Tumoren, die im Verlauf entstehen können, sind beispielsweise: Optikusgliom, pilozytisches Astrozytom, Phäochromozytom und Rhabdomyosarkom. Entwicklungsverzögerungen und Lernschwierigkeiten treten bei etwa der Hälfte der Betroffenen auf. Der Kopfumfang liegt häufig im oberen Normbereich oder im Bereich der Makrozephalie. Bei zwei von drei Kindern mit einer NF1 finden sich in der craniellen MRT (T2-Wichtung) sogenannte FASI (Foci mit abnormer Signal-Intensität, Abb. 6). Diese entsprechen Hirnarealen mit spongiformer Myelopathie, ihre Bedeutung ist jedoch bislang unklar (Friedman, 2012).
Diagnostische Kriterien
Die klinische Diagnosestellung einer NF1 erfolgt anhand von Kriterien, von denen zwei oder mehr erfüllt sein müssen. Allerdings entwickeln sich diese Symptome meist erst im Lauf der ersten Lebensjahre. Nur etwa die Hälfte der Kinder, deren Eltern nicht von einer NF1 betroffen sind, erfüllen die klinischen NF1-Kriterien während des ersten Lebensjahres, im Alter von acht Jahren sind dagegen nahezu alle Kinder ohne familiäre Vorgeschichte klinisch zu diagnostizieren (Friedmann und Birch, 1997; DeBella et al., 2000).
Bei Kindern mit einem betroffenen Elternteil lässt sich die Diagnose meist bereits innerhalb des ersten Lebensjahres stellen, da zusätzlich zur positiven Familienanamnese nur ein weiteres Kriterium notwendig ist. Dieses zusätzliche Kriterium ist bei den allermeisten Kindern das Auftreten von Café-au-lait Flecken (DeBella et al., 2000; Nunley et al., 2009).

Genetische Ursachen der Neurofibromatose Typ 1
Die Neurofibromatose Typ 1 wird durch Mutationen im NF1-Gen verursacht. Das NF1-Gen liegt auf dem langen Arm von Chromosom 17 (17q11.2) und kodiert für das Protein Neurofibromin. Dieses Protein reguliert das Zellwachstum und die Zelldifferenzierung in verschiedenen Geweben, zum Beispiel in Nervenzellen und deren „Schutzhülle“ (Myelinscheide). Bei Mutationen im NF1-Gen (Tumorsuppressorgen) kann es daher zu übermäßigem Zellwachstum und somit zur Tumorentstehung kommen, zum Beispiel zum Auftreten von Neurofibromen. Bei fast allen klinisch Betroffenen mit einer Neurofibromatose Typ 1 lassen sich Mutationen des NF1- Gens nachweisen (90 Prozent Punktmutationen; etwa 6 Prozent Deletionen). Die genetische Diagnostik kann aus einer Blutprobe erfolgen und ist Leistung der gesetzlichen Krankenversicherung (siehe: Infobox Seite 10). Die Vererbung der NF1 erfolgt autosomal-dominant. Dies bedeutet, dass Betroffene die ursächliche NF1- Mutation an durchschnittlich die Hälfte ihrer Kinder weitergeben. Dabei ist jedoch die variable Expressivität von NF1-Genmutationen zu beachten: Manche NF1-Genträger weisen nur sehr milde Symptome auf (z. B. ausschließlich Café-au-lait Flecken und Freckling), andere können jedoch sehr stark betroffen sein (z. B. multiple Neurofibrome, intracranielle Tumore). Der relativ hohe Anteil von 50 Prozent Neumutationen im NF1-Gen hängt damit zusammen, dass das NF1-Gen vergleichsweise groß (60 Exons) und somit „anfällig“ für Mutationen ist.
Klinisches Management bei einer Neurofibromatose Typ 1
Bei einer Neurofibromatose Typ 1 wird das folgende klinische Follow-up empfohlen (bei Kindern halbjährlich, bei Erwachsenen jährlich):
- Klinisch-neurologische Untersuchung
- Dokumentation somatischer Daten (Kopfumfang häufig erhöht)
- Augenärztliche Untersuchung einschließlich Perimetrie und Funduskopie
- Blutdruckmessung